Cell Mixture Analysis using Seq-Well Sequencing
SW_CellMixture_SIGURD.RmdCell Mixture Analysis using SIGURD
In this data set, chronic myelogenous leukemia cells (K562) and brain tumor cells (BT142) were mixed and then sequenced using Seq-Well S3. The cells can be separated using the expression of marker genes.
The original script then identifies 6 variants that can distinguish between the two cell types.
In this reproduction, we show that the analysis can be faithfully reproduced using a very streamlined set of functions. This greatly simplifies the analysis. Using a set of dedicated functions, the analysis can now also be easily adopted to new data sets.
Loading necessary packages.
We load the data from MAESTER.
print("Libraries for SIGURD.")## [1] "Libraries for SIGURD."We load the Seurat object for the scRNA-seq data.
seu <- readRDS("/data/MPN/exp/scRNA/MPN_mutations/SIGURD_paper/sigurd/data/SW_CellLineMix_Seurat_Keep_Renamed.rds")Loading the data using SIGURD.
SIGURD loads the data and automatically reformats the data. This code uses dedicated functions to load the and process the data, which streamlines the analysis quite substantially.
# The design matrix contains information for the genotyping data.
genotyping <- LoadingMAEGATK_typewise(patient = "SW", samples_file = "/data/MPN/exp/scRNA/MPN_mutations/SIGURD_paper/sigurd/data/MAESTER_Reproduction.csv", min_cells = 0, type_use = "Amplicon_MT", verbose = FALSE)
# Removing cells that are not in the Seurat object.
genotyping <- Filtering(genotyping, cells_include = colnames(seu))## [1] "We remove all cells not in the allow list."
## [1] "We remove all the variants that are always NoCall."
# Adding meta data do the SIGURD object.
colData(genotyping)$CellType <- seu$CellTypeSelecting the Variants of Interest
Now, we select the variants of interest.
# Selecting informative variants.
voi.ch.sigurd <- VariantSelection_Quantile(genotyping, min_coverage = 20, min_quality = 30, quantiles = c(0.1, 0.9), thresholds = c(0.1, 0.9), verbose = FALSE)
print(voi.ch.sigurd)## [1] "chrM_709_G_A" "chrM_1888_G_A" "chrM_1420_T_C" "chrM_2141_T_C"
## [5] "chrM_9117_T_C" "chrM_7990_C_T"Visualisation using SIGURD
# Determining if a cell is supporting one of the two cell types.
cell_support <- CallSupport(SE = genotyping, VOI_group1 = voi.ch.sigurd[voi.ch.sigurd != "chrM_7990_C_T"], VOI_group2 = "chrM_7990_C_T", group1_name = "K562", group2_name = "BT142", min_mutated_reads = 3, min_reads = 30, group_factor = 10, verbose = FALSE, return_nonsupport = TRUE)
colData(genotyping)$CellType_MT <- cell_support$Support
cell_support <- subset(cell_support, Support %in% c("K562", "BT142")) # Selecting only cells with sufficient support for a set of variants.
# Plotting the results on a heatmap.
HeatmapVoi(SE = genotyping[,cell_support$Cell], voi = voi.ch.sigurd, annotation_trait = "CellType", minimum_coverage = 3, remove_empty_cells = FALSE)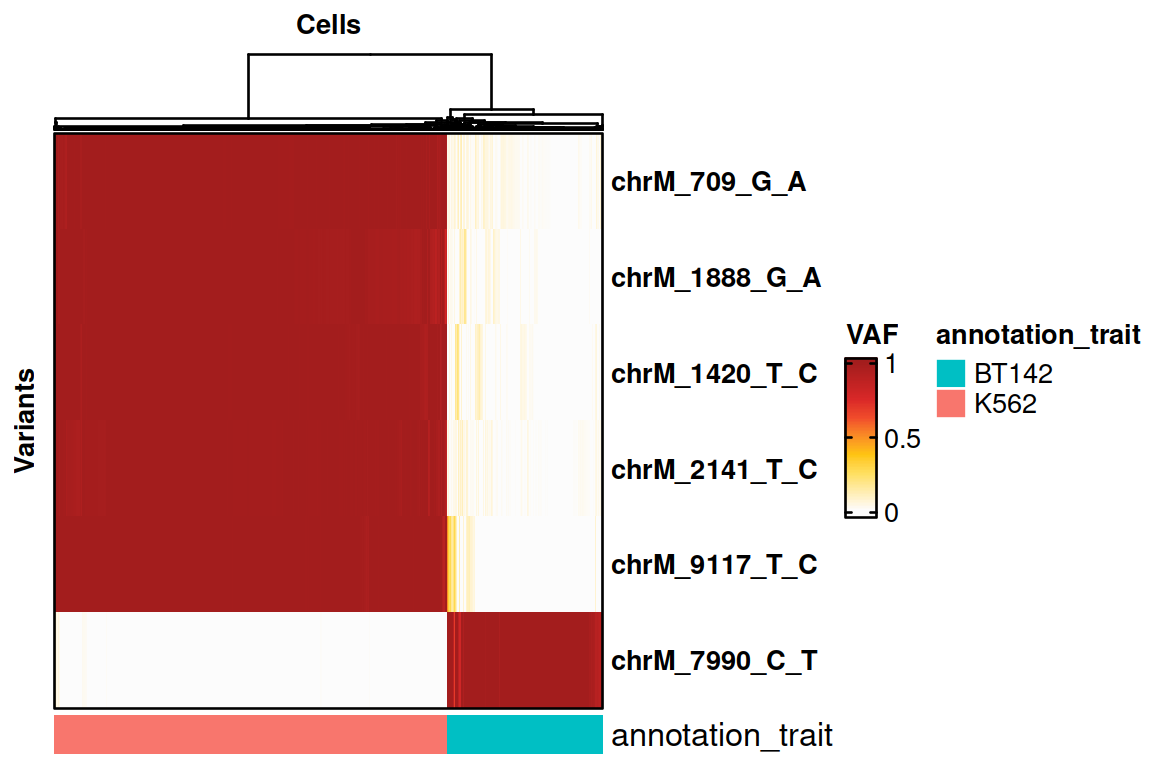
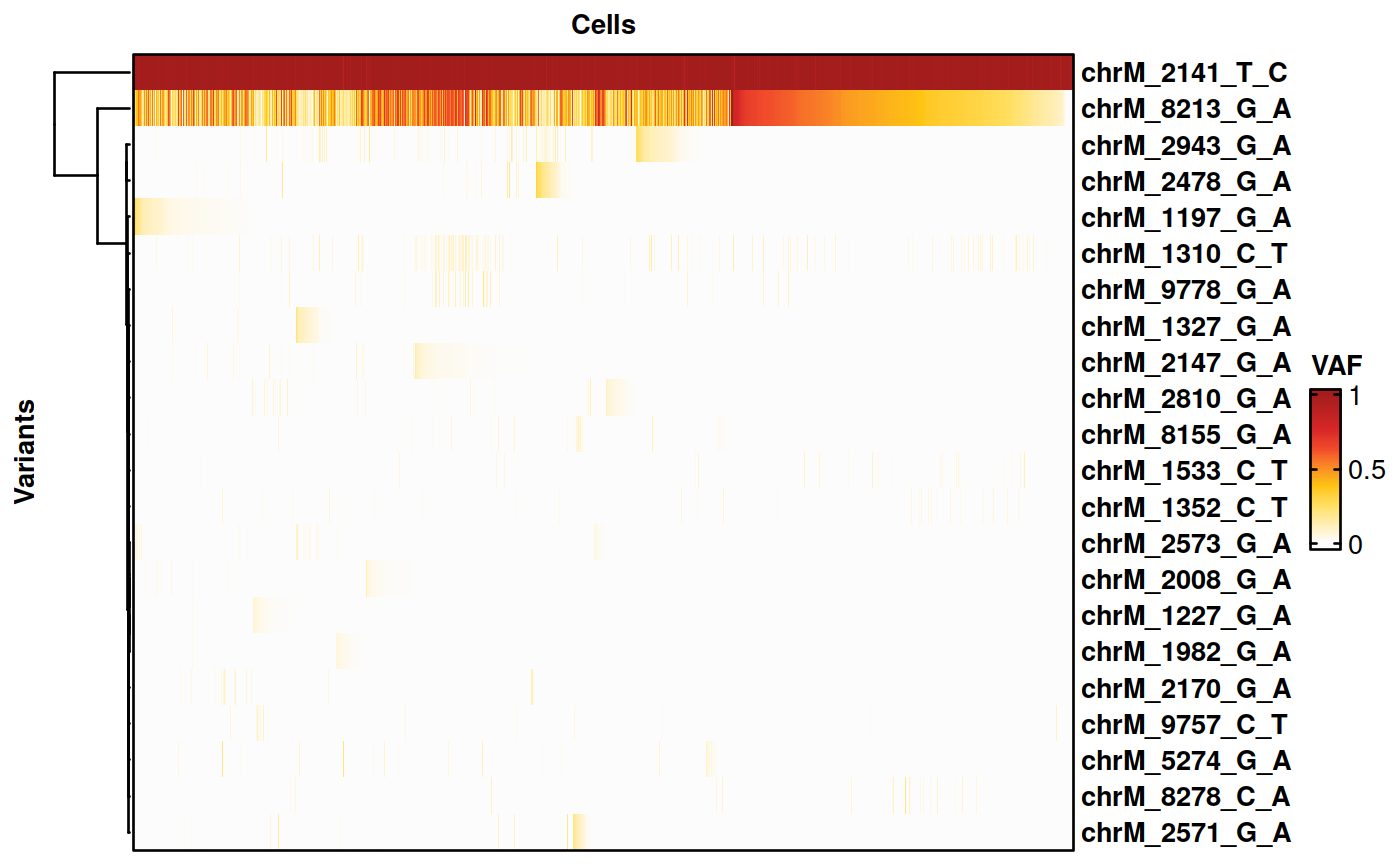
Selecting variants with comparatively high VAF between groups
We now select variants with a higher VAF in the K562 cells than in the BT142 cells.
voi.ch.lineages <- sigurd::VariantSelection_Group(SE = genotyping, min_coverage = 20, quantiles = c(0.01, 0.99), thresholds = c(0.01, 0.02), min_quality = 30,
group_of_interest = "CellType_MT", group1 = "K562", group2 = "BT142", group_factor = 5, remove_nocall = FALSE, verbose = FALSE)
voi.ch.lineages <- gsub("chrM_9117_T_C", "chrM_2141_T_C", voi.ch.lineages) # Replace chrM_9117_T_C, which comes up because it has no coverage in some cells, with another homoplasmic variant (chrM_2141_T_C), that has better coverage)
voi.ch.lineages <- voi.ch.lineages[!voi.ch.lineages %in% c("chrM_5378_A_G", "chrM_6384_G_A")] # These variants are not detected in the bulk data and were removed by hand.
print(voi.ch.lineages)## [1] "chrM_1197_G_A" "chrM_1227_G_A" "chrM_1327_G_A" "chrM_1982_G_A"
## [5] "chrM_2008_G_A" "chrM_2147_G_A" "chrM_2170_G_A" "chrM_2478_G_A"
## [9] "chrM_2571_G_A" "chrM_2573_G_A" "chrM_2810_G_A" "chrM_2943_G_A"
## [13] "chrM_5274_G_A" "chrM_8155_G_A" "chrM_8213_G_A" "chrM_8278_C_A"
## [17] "chrM_9778_G_A" "chrM_2141_T_C" "chrM_1310_C_T" "chrM_1352_C_T"
## [21] "chrM_1533_C_T" "chrM_9757_C_T"Visualisation using SIGURD
# Plotting the results on a heatmap.
cell_support <- subset(cell_support, Support == "K562")
genotyping <- Filtering(genotyping, cells_include = cell_support$Cell, verbose = FALSE)
HeatmapVoi(SE = genotyping, voi = voi.ch.lineages, minimum_coverage = 3, sort_cells = TRUE, cluster_variants = TRUE)